钴催化高区域选择性内炔硼氢化反应
南开大学化学学院朱守非课题组发展了一类新型环丙烷骨架双膦配体-钴催化剂,实现了非对称内炔的高区域选择性硼氢化反应:当以芳基烷基内炔作为底物时,可获得优异的β-加成选择性;而以双烷基内炔为底物时,则获得很高的α-加成选择性——这是其它已知催化剂无法实现的。该反应条件温和,操作简单,易于放大,产物易于转化,具有很好的合成应用前景,为构型确定的三取代烯基硼和三取代烯烃的合成提供了新方法。机理实验表明起始的活性催化剂可能是Co(I)-H物种,负氢从金属到炔烃底物的不可逆转移是该反应的区域选择性决定步。刚性的环丙烷骨架配体及其与金属组成的拥挤空腔是取得高选择性的决定性因素。该研究表明新配体的发展将给3d金属催化带来新的机遇。该成果近期发表于Angew. Chem. Int. Ed.(https://doi.org/10.1002/anie.202208473),博士生张艳东是文章的第一作者。
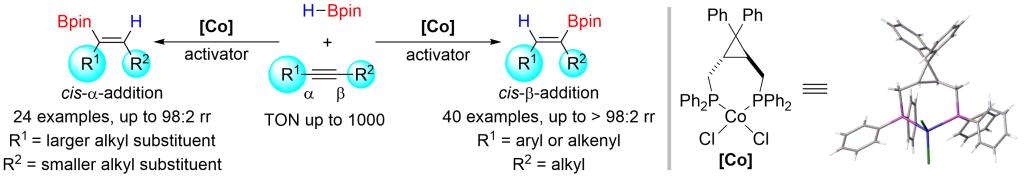
图1. 钴催化高区域选择性内炔硼氢化反应
有机硼化合物是分子内含有C-B键化合物的统称,是一类重要的功能分子,被广泛应用于有机合成,医药及材料等领域。作为多样化的合成砌块,烯基硼化合物被广泛应用于Suzuki偶联,Chan-Lam偶联,Petasis反应等,可以实现由C-B到C-C及C-X (X = N, O, S, Cl, Br, I)等一系列转化。炔烃硼氢化是合成烯基硼化合物的有效方法,具有原料来源广泛、反应条件温和可控、原子经济性高等特点。相较于端炔,内炔位阻大,反应活性低,其硼氢化还存在化学、区域和立体选择性难以调控的问题。因此发展用于非对称内炔硼氢化反应的新型过渡金属催化剂,特别是基于丰产金属的催化剂,实现文献中还不能很好控制的选择性,具有重要的研究价值。
由于3d金属(即第一过渡系金属)储量丰富、廉价易得、生物兼容性好,近年来基于 3d金属催化剂的研究受到广泛关注。南开大学朱守非课题组长期致力于3d金属催化的反应研究(Angew. Chem. Int. Ed. 2014, 53, 13188; J. Am. Chem. Soc. 2017, 139, 3784; J. Am. Chem. Soc. 2017, 139, 7697; Chem. Sci. 2017, 8, 7197; Nat. Commun.2018, 9, 221; J. Am. Chem. Soc. 2018, 140, 7458; Science 2019, 366, 990; J. Am. Chem. Soc. 2019, 141, 4579; Org. Lett. 2019, 21, 7883; J. Am. Chem. Soc. 2020, 142, 16894; J. Am. Chem. Soc. 2021, 143, 6962; CCS Chem. 2021, 3, 1721; J. Am. Chem. Soc. 2022, 144, 515; Angew. Chem. Int. Ed. 2022, e202203343; ACS Catal. 2022, 12, 2581; Chem. Sci. 2022, 13, 2721; Chem. Sci. 2022, 13, 7873),在此基础上该课题组报道一类新型的环丙烷骨架双膦配体-钴催化剂,可以实现内炔的高区域选择性硼氢化反应:对于芳基烷基内炔给出优异的β-加成选择性(硼加成至烷基一端,up to > 98:2 rr);而对于非对称双烷基内炔,则给出了罕见的α-加成选择性(硼加成至大位阻烷基一端,up to 98:2 rr)。

图2. 非对称内炔的硼氢化反应
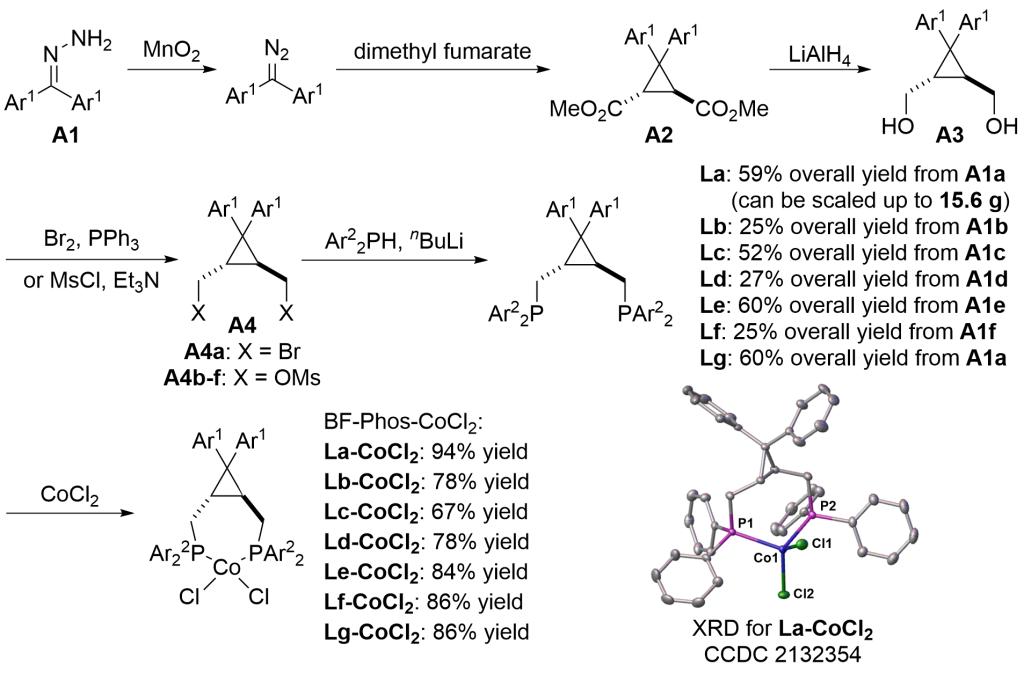
该工作中使用的最优配体BF-Phos(因单晶显示其结构类似蝴蝶,故名Butterfly Phos,缩写为BF-Phos)为一类新型配体,以市售的二苯基甲酮腙为起始原料,经过5步反应即可以64%总收率制得。将BF-Phos与CoCl2络合即可获得性质稳定的蓝色粉末,单晶结构显示BF-Phos作为双齿配体与CoCl2螯合配位,钴原子周围的四个配位原子组成四面体结构,P-Co-P的咬合角为109˚。
Selected bond lengths [Å] and angles [°]: Co1-P1 2.3701(5), Co1-P2 2.3632(5), Co1-Cl1 2.2204(5), Co1-Cl2 2.2068(5); P1-Co1-P2 108.997(19), Cl1-Co1-Cl2 117.22(2). (Hydrogen atoms and solvent, DCM were omitted for clarity.)
图3. BF-Phos-CoCl2的合成及单晶结构表征
该BF-Phos-CoCl2配合物可以催化内炔的硼氢化。对于芳基烷基内炔均能够以中等至优异的收率(54-99% yields)及良好的区域选择性(91:9 to >98:2 rr)获得三取代烯基硼酸酯产物。对于共轭烯炔,能够以良好的收率和选择性得到共轭二烯基硼酸酯(图4)。
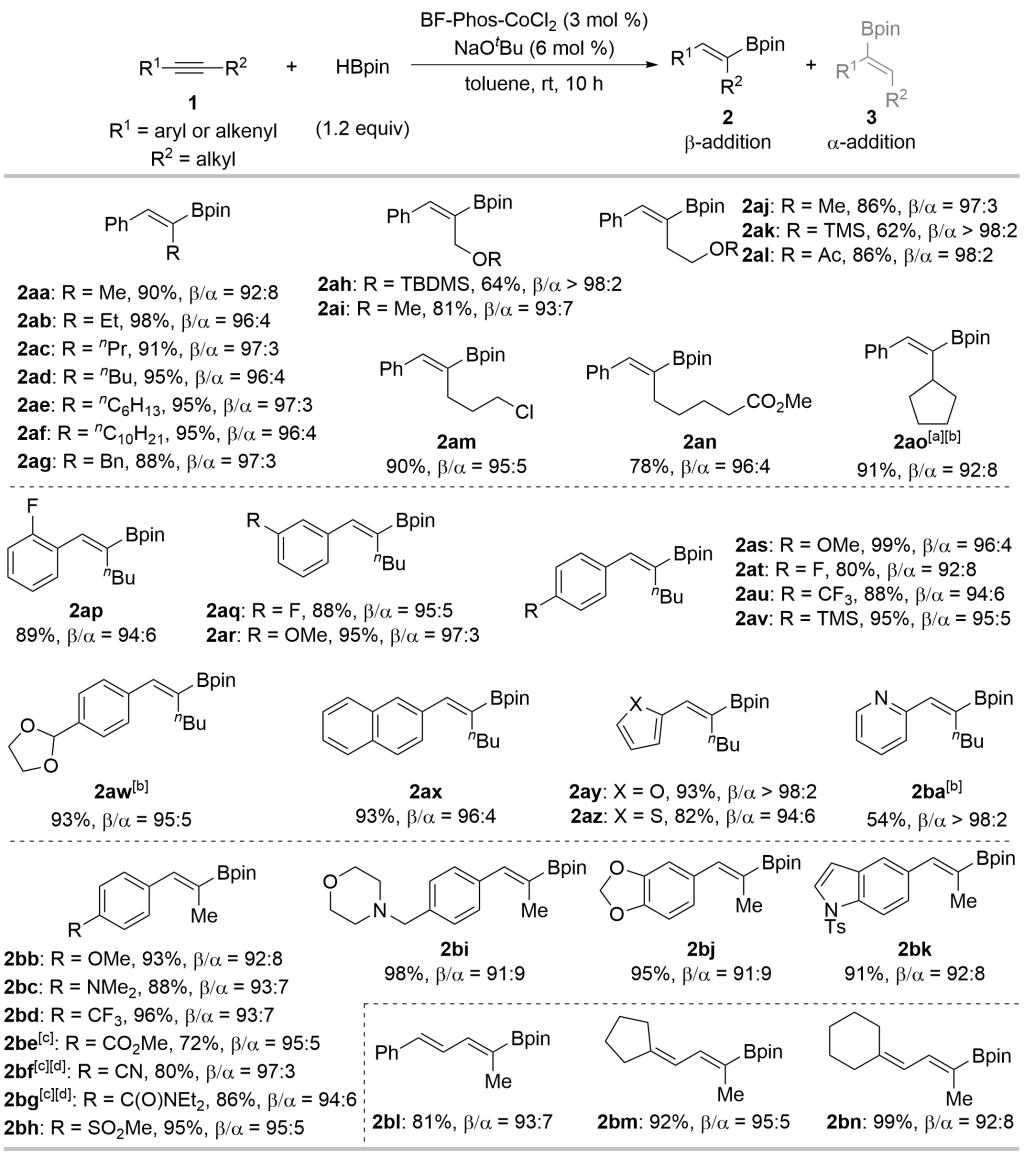
Reaction conditions: 1 (0.5 mmol), HBpin (0.6 mmol, 1.2 equiv), La-CoCl2 (3 mol %), and NaOtBu (6 mol %) in toluene (2 mL) were stirred at rt for 10 h. Isolated yields were given. The regioisomeric ratios (rr, β/α) were determined by 1H NMR. [a] The reaction was performed at 0.2 mmol scale with Lg-CoCl2 as catalyst. [b] Reaction time: 24 h. [c] Reaction time: 12 h. [d] Used 0.55 mmol HBpin (1.1 equiv).
图4. 钴催化芳基烷基内炔硼氢化底物适用范围
催化剂BF-Phos-CoCl2在双烷基内炔硼氢化反应中,可以将硼基团选择性地加成至大位阻的烷基一端,并且表现出广泛的底物适用范围(图5)。即便对于两端取代基差异很小的2-戊炔,该催化剂依然能够区分乙基与甲基,以很高的区域选择性(91:9 rr)得到α-加成产物。在不对称双烷基内炔的硼氢化反应中,以如此高的区域选择性(>10:1 rr)获得α-加成产物是未见报道的,而由此获得的构型确定的三取代烯基硼酸酯也难以通过其它方法合成。
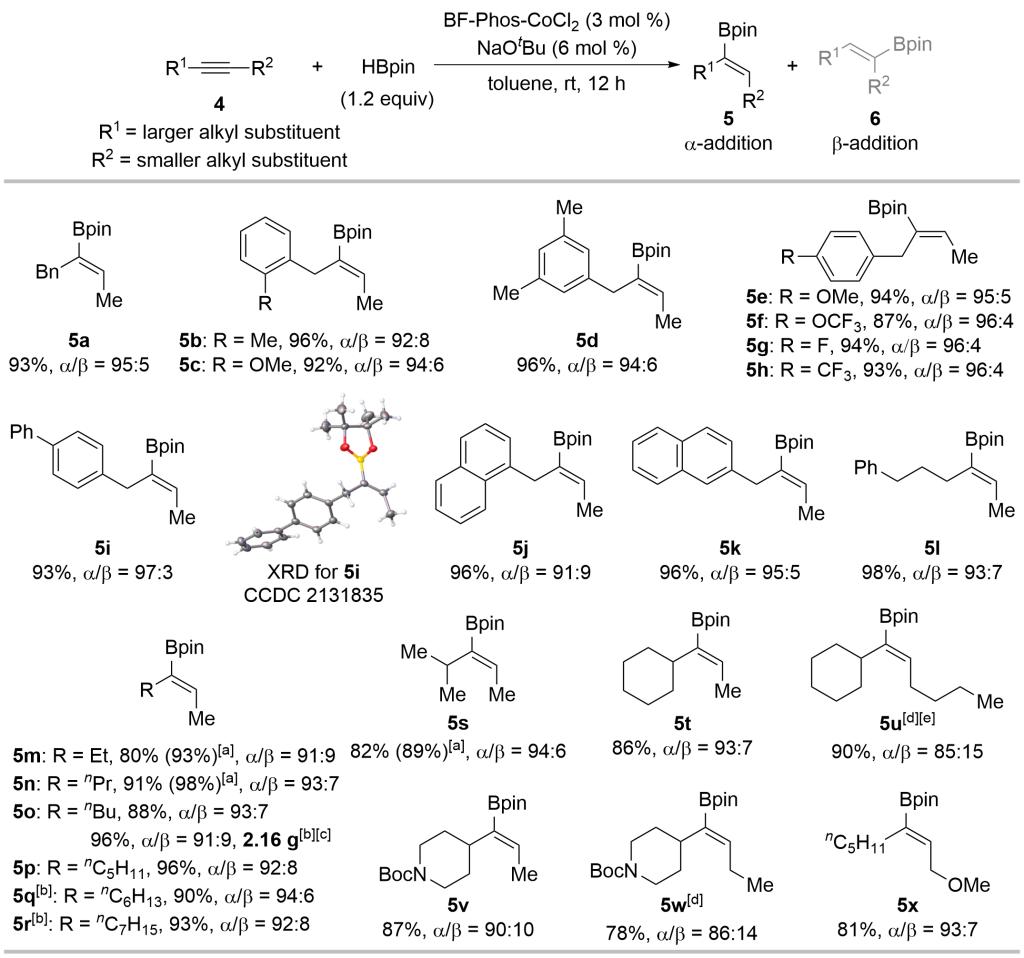
Reaction conditions: 4 (0.5 mmol), HBpin (0.6 mmol, 1.2 equiv), La-CoCl2 (3 mol %), and NaOtBu (6 mol %) in toluene (2 mL) were stirred at rt for 12 h. Isolated yields were given. The regioisomeric ratios (rr, α/β) were determined by 1H NMR. [a] Yields determined by 1H NMR using CH2Br2 as an internal standard were given in parentheses. [b] Used 1 mol % BF-Phos-CoCl2 and 2 mol % NaOtBu. [c] Used 10 mmol 4o. [d] Reaction time: 24 h. [e] Used 0.4 mmol 4u.
图5. 钴催化双烷基内炔硼氢化底物适用范围
双烷基内炔硼氢化产物是一类新型的烯基硼酸酯,其C-B键可以构型保持地转化为转化为C-C键及C-X (X = N, O, Cl, Br, I)键,这表明该研究发展的方法学在三取代烯烃的合成中具有很好的应用潜力(图6)。除此之外,研究者还将该硼氢化反应成功应用于一系列生物活性分子(如替马罗汀、芳维甲酸等)及天然产物(咔唑生物碱)关键中间体的合成中,显著提高了合成效率。

Reaction conditions: (a) methyl 4-bromobenzoate, Pd(PPh3)4, Cs2CO3, THF, 65 ˚C, 12 h; (b) ethyl acrylate, Pd(OAc)2, 1,10-phenanthroline, O2 balloon, N,N-dimethylacetamide, 80 ˚C, 12 h; (c) methyl vinyl ketone, [Rh(COD)Cl]2, K3PO4, 1,4-dioxane/H2O, 80 ˚C, 12 h; (d) ICH2Cl, nBuLi, THF, -78 ˚C to rt, 10 h; then NaOH (aq., 2M), 30% H2O2, 0 ˚C, 2 h; (e) NaOH (aq., 2M), 30% H2O2, THF, 0 ˚C, 3 h; (f) for chlorination: CuCl2, THF/MeOH/H2O, 100 ˚C, 36 h; for bromination: CuBr2, EtOH/H2O, 100 ˚C, 24 h; for iodination: KI, CuI, 1,10-phenanthroline, MeOH/H2O, 80 ˚C, 12 h; (g) NaN3, CuSO4, MeOH, 50 ˚C, 20 h.
图6. 三取代烯基硼酸酯的克级规模合成及转化
为了揭示可能的反应机理,研究者进行了一系列控制实验(图7)。当量还原实验显示活性催化剂可能为Co(I)-H物种(图7a);所得的活性催化剂A在内炔的硼氢化反应中给出与标准条件下类似的反应结果(图7b);活性催化剂A的1H NMR谱图中可以观察到负氢信号;XPS测试表明活性催化剂A中钴的价态可能为一价。为了进一步确认活性催化剂A的组成(Co-H or Co-Bpin),研究者使用炔烃底物与当量的活性催化剂A混合,之后以甲醇淬灭反应,结果可以观测到烯烃的产生,这说明活性催化剂A应为含Co-H的物种(图7c)。KIE实验没有观察到明显的动力学同位素效应(图7d),表明氢转移过程可能并不包含在决速步中。

图7. 机理实验
根据上述控制实验,参考过往文献报道,研究者提出了图8所示的催化循环:首先钴催化剂前体BF-Phos-CoCl2在叔丁醇钠与HBpin共同作用下生成活性催化物种Co(I)-H A,随后与炔烃配位,迁移插入得到烯基钴中间体C,之后与HBpin发生σ键复分解生成中间体E,并通过配体交换生成烯基硼酸酯产物,完成催化循环。

图8. 可能的催化循环
为了阐明区域选择性的来源,研究者对可能的反应路径进行了DFT计算。计算结果表明Co-H对炔烃底物的迁移插入步骤为不可逆过程,决定了反应的区域选择性。相应主副产物过渡态的能量差符合实验事实。此外,研究者还通过独立梯度模型(IGM)分析进一步解释了主副产物过渡态能量差异的来源(π–π及C–H···π相互作用),以及通过键角及二面角的分析讨论了主要产物过渡态能量较低的合理性。研究者认为选择性的来源可追溯到催化剂的结构与性质:作为第一过渡系金属,钴原子半径小,具有独特的开壳层结构;作为最小的环系结构,环丙烷结构刚性,环上取代基相互影响;二者搭配组成的催化剂,形成了拥挤的反应口袋,从而能够精准识别炔烃两端取代基的微小差异,给出优异的区域选择性。
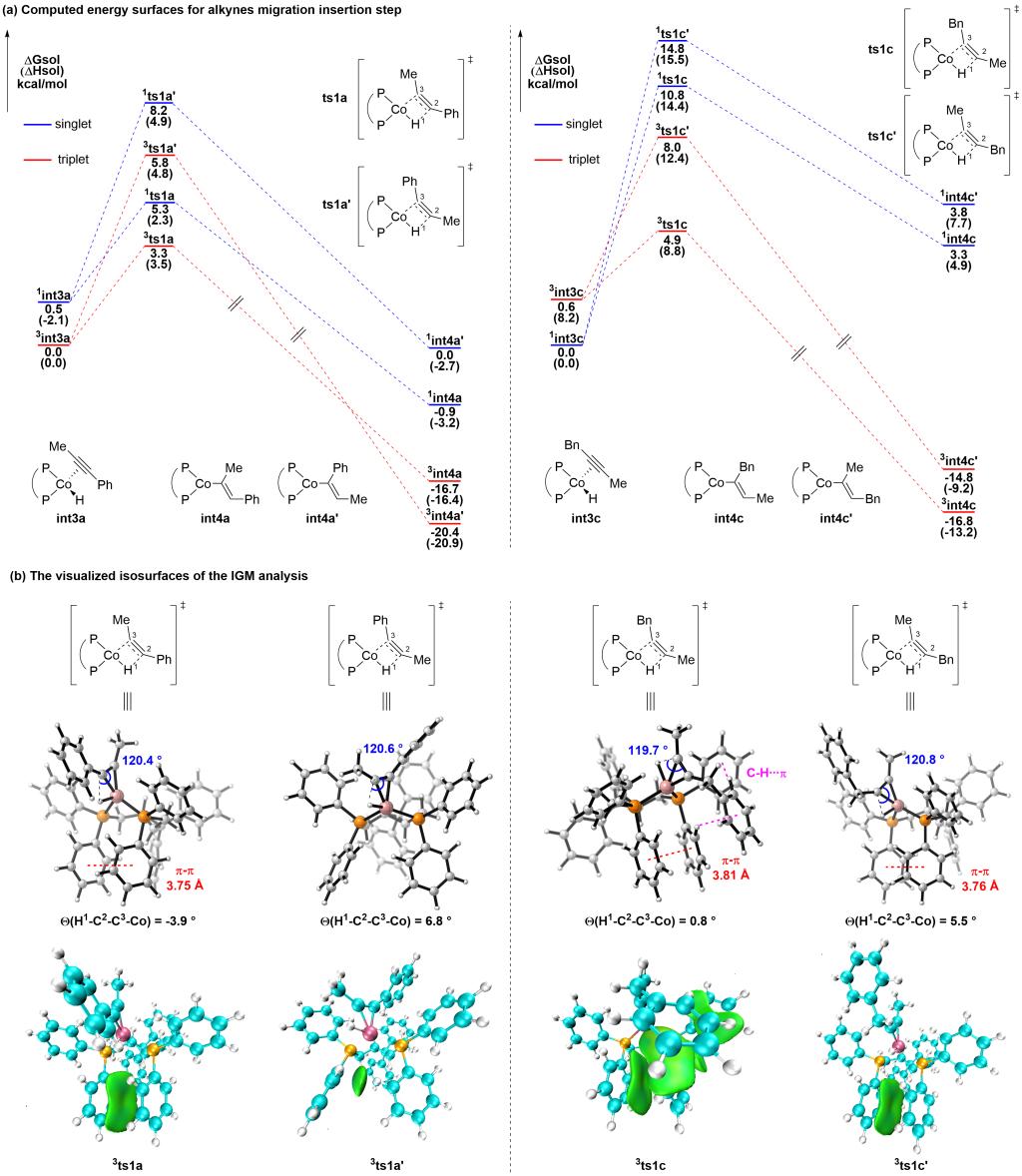
图9. DFT计算
总之,朱守非课题组发展了一类基于环丙烷骨架双膦配体的钴催化剂,实现了非对称内炔的高化学、区域、立体选择性硼氢化反应。该方法具有广泛的底物适用范围,可以兼容多种官能团(酯基,氰基,酰胺,磺酰基等)和杂环(呋喃,噻吩,吲哚,吡啶等)结构。产物三取代烯基硼酸酯中的C-B键可以构型保持地转化为C-C键及C-X(X = N, O, Cl, Br, I)键,被成功应用于一系列生物活性分子及天然产物中间体合成,显著提升了合成效率。机理研究表明,新型环丙烷双膦配体和钴搭配形成的开壳层催化剂具有拥挤的反应空腔,这可能是反应获得高选择性的关键。该工作拓展了钴催化硼氢化反应的边界,深化了开壳层催化剂性质的理解,为三取代烯基硼酸酯及烯烃的合成提供了新的有效途径。
