Research: Cobalt-Catalyzed Hydrosilylation of 1,3-Enynes for the Synthesis of Conjugated Alkenylalkoxysilanes
钴催化1,3-烯炔硅氢化反应合成共轭烯基烷氧基硅烷
背景介绍
共轭烯基烷氧基硅烷在有机合成与功能材料领域具有重要应用价值,可用于聚合物交联网络构建、界面功能修饰及复合工程材料设计。1,3-烯炔与烷氧基硅烷的硅氢化反应是构建该类化合物的高效方法,兼具反应条件温和、原子经济性高、易于规模化制备等优势。但1,3-烯炔反应位点多、反应活性高,化学选择性、区域选择性及立体选择性精准调控难度大(图1A)。
目前1,3-烯炔与伯、仲硅烷的硅氢化已有较多催化体系报道;对于极性酯基取代1,3-烯炔与叔硅烷的反应,也可通过铑、铂催化剂实现高选择性合成(图1B)。然而非官能团化1,3-烯炔与叔烷氧基硅烷的高选择性硅氢化仍存在很大挑战。本研究采用环丙烷稠合双膦配体配位的钴催化剂,实现了非官能化1,3-烯炔与叔烷氧基硅烷的高效硅氢化反应。具体而言,芳基膦配体可促使芳基取代1,3-烯炔发生高选择性顺式-α-硅氢化反应(收率最高 95%,区域选择性>95:5);与之相反,环己基膦配体能够促进烷基取代1,3-烯炔发生顺式-β-硅氢化反应(收率最高 99%,区域选择性>95:5)。该反应体系官能团兼容性优异,可便捷放大至克级制备规模,能够高效合成一系列结构新颖的共轭烯基烷氧基硅烷。所得产物可进一步转化为有机硅材料前驱体,为高端功能性有机硅新材料的研发提供新的物质基础。
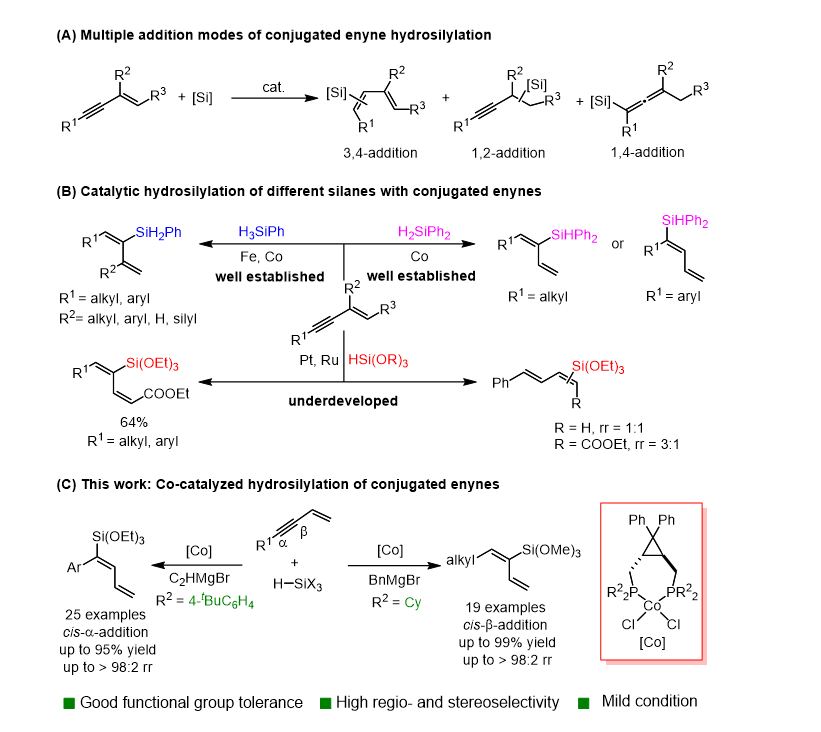
图1. 研究背景
反应条件优化
研究首先以芳基取代 1,3 - 烯炔与三乙氧基硅烷的硅氢化作为模板反应,筛选配体、钴盐、活化剂及溶剂体系。最终确定最优条件:以环丙烷并双膦配体配位钴配合物 L2-CoCl2为催化剂,乙炔基溴化镁为活化剂,甲苯为溶剂室温反应,以 > 98% 收率、>95:5 高区域选择性专一得到顺式-α-加成产物。
配体筛选实验表明:环丙烷骨架结构对提升双膦配体区域调控能力至关重要;环丙烷骨架上双联芳基取代可通过位阻效应提升催化剂稳定性;文献适用于1,3 -烯炔与仲硅烷硅氢化的 dppf、Xantphos 等配体,在烷氧基硅烷体系中催化性能极差,表明叔硅烷与仲硅烷存在显著反应活性差异。
针对烷基取代1,3-烯炔,进一步筛选得到环己基膦修饰的钴配合物 L3-CoCl2催化体系,以苄基溴化镁为活化剂、环己烷为溶剂,高效实现顺式-β-选择性硅氢化,区域选择性可达 >95:5。
底物范围
芳环邻、间、对位取代的芳基1,3-烯炔均能保持高活性与高区域选择性;芳环给电子/吸电子取代基对反应影响较小,缺电子底物区域选择性更优异。反应官能团兼容性优异,炔基、缩醛、硅基等敏感官能团均可耐受;杂环、萘基、环己烯基取代1,3-烯炔以及双烯炔结构底物均能顺利反应,以 73%–92% 收率、≥95:5 区域选择性得到目标产物(图2)。
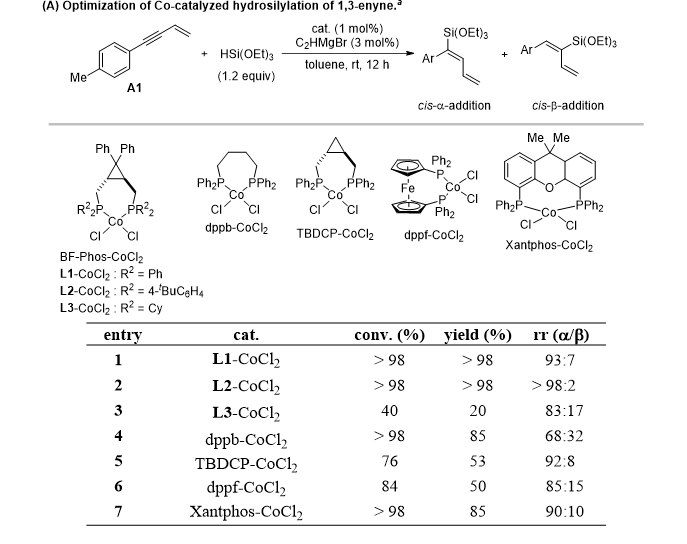
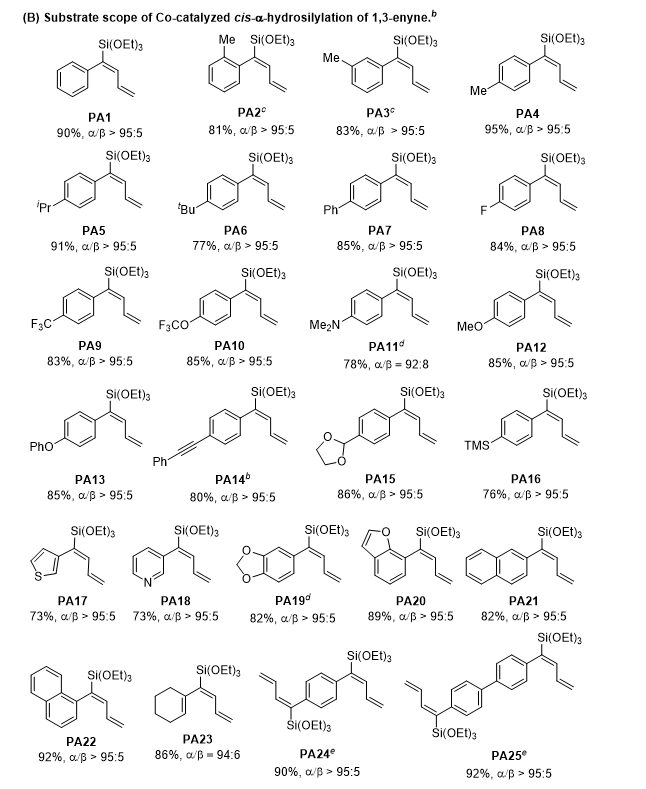
图2. 芳基取代1,3-烯炔底物的反应结果
不同烷基取代的1,3-烯炔和氯/硫/醚等杂原子官能化烷基1,3-烯炔均适用,收率74%–95%、区域选择性≥92:8;各类苄氧基取代底物、仲碳烷基取代底物均表现优异,烷基位阻增大可进一步提升区域选择性;烯基位点取代底物也能以 90% 收率、>95:5 区域选择性完成转化(图3)。
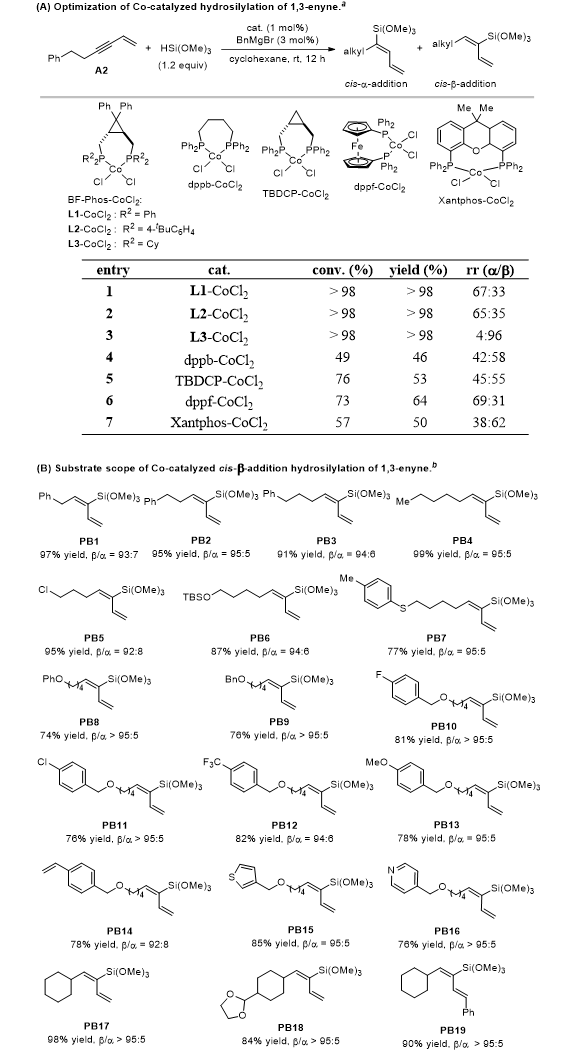
图3. 烷基取代的1,3-烯炔底物的反应结果
硅烷试剂适用范围
三取代烷氧基硅烷中,烷氧基数目越多Si-H键极性越强、反应活性越高;位阻较大的三异丙氧基硅烷活性显著降低。该催化体系对二苯基硅烷、苯基硅烷同样兼容,且活性与选择性优于文献已知催化体系(图4)。
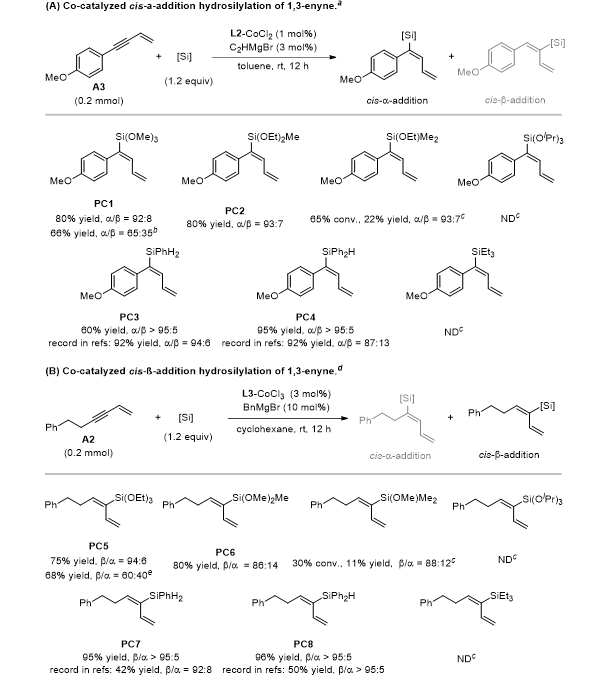
图4. 硅烷试剂适用范围
产物转化与合成应用
该钴催化体系可顺利实现克级放大合成,8 mmol 规模下仍保持≥90% 分离收率与优异区域选择性,具备实际制备价值(图5A)。产物可通过配体交换转化为稳定性更高的硅烷衍生物,亦可构筑硅烷交联聚乙烯关键骨架新型共轭烯基硅化合物(图5B);同时可经水解、缩聚制备含共轭二烯结构的烷氧基硅烷聚合物,为高性能硅油、硅烷偶联剂及有机硅功能高分子提供重要前驱体(图5C)。
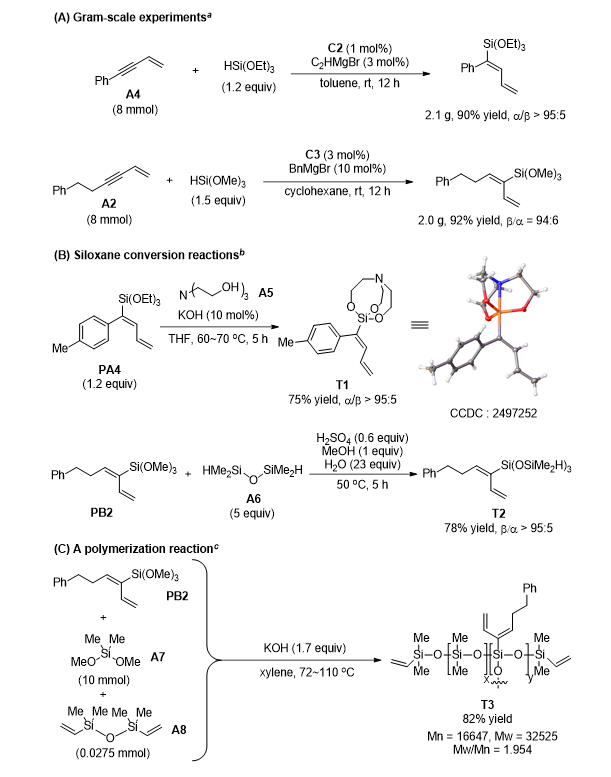
图5. 产物转化
机理实验
自由基捕获实验排除自由基反应路径;硅烷交叉对照实验证明一分子硅烷完整参与加成,不存在 Co-H/Co-Si 中间体分子间氢硅迁移。成功制备明确结构 L1-Co (0) 配合物,无需格氏试剂活化即可催化反应,证实Co(0)为真实活性物种;格氏试剂通过金属转移-还原消除过程原位生成低价Co(0)活性中心。动力学同位素效应(KIE)实验显示两种选择性体系 KIE 值均接近 1,表明Si-H 键断裂不参与决速步。
反应的可能机理是:Co(0) 先与 1,3-烯炔炔基配位,再活化硅烷 Si-H 键,经配体到配体的氢转移(LLHT),最后还原消除得到目标产物;芳基膦配体导向α-加成,环己基膦配体导向β-加成。
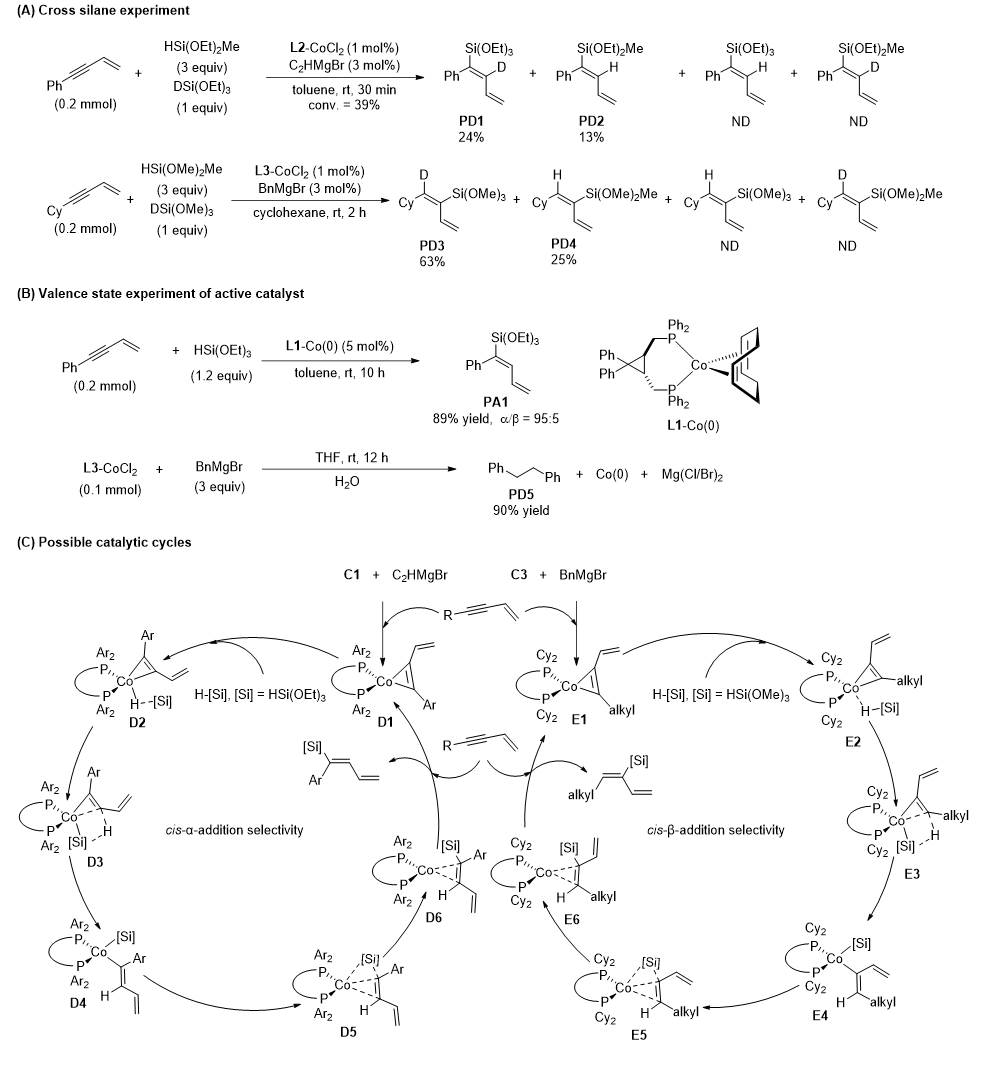
图6. 机理实验
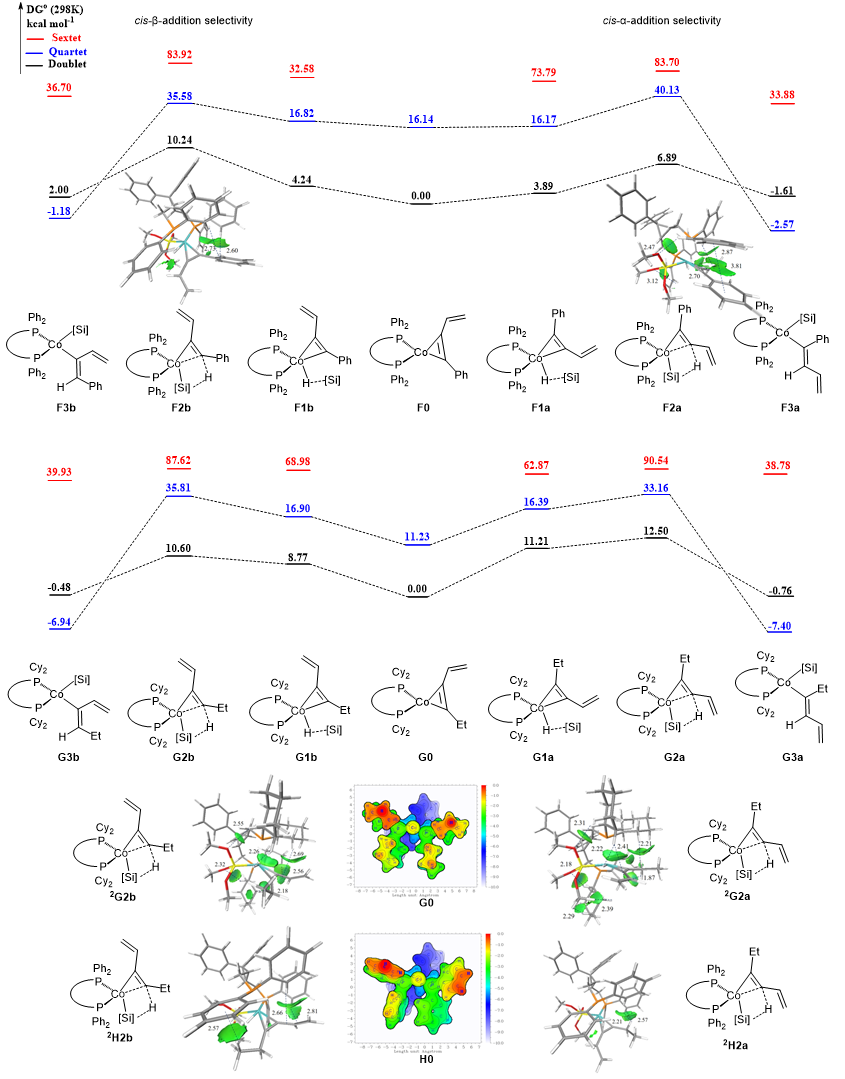
DFT 计算及选择性起源
DFT 计算表明:芳基膦-钴催化体系中,顺式-α-加成过渡态能垒比 β 路径低 3.0 kcal・mol-1,与实验的 α 选择性一致;IGMH 分析证实过渡态中π-π 堆积作用显著稳定中间体结构,降低反应能垒。
烷基 1,3-烯炔体系中,环己基膦配体催化下 β 路径能垒低 1.9 kcal・mol-1;电荷布居分析显示氢迁移方向差异造成过渡态静电作用强弱不同,决定 β 选择性优势。空间结构与弱相互作用分析表明:环己基膦配体位阻更强、给电子能力更显著,钴中心电子更富集,可通过范德华作用与底物形成丰富弱相互作用,精准识别微小位阻差异的烷基烯炔底物;而芳基膦主要依靠 π-π 堆积实现选择性调控。两类配体空间构型相近,但位阻、电子效应与非共价作用互补,实现了底物类型匹配的区域发散调控。
结论
南开大学朱守非课题组开发了系列环丙烷并双膦配体调控的钴催化体系,首次实现非官能团化 1,3 - 烯炔与叔烷氧基硅烷的高效区域发散式硅氢化。通过配体电子效应与空间位阻调控,分别实现芳基 1,3 - 烯炔高选择性顺式-α-硅氢化、烷基1,3-烯炔顺式-β-硅氢化,收率最高达 99%、区域选择性普遍 > 95:5。反应官能团耐受性好、可克级放大,产物可衍生为有机硅材料前驱体与功能聚合物。结合控制实验与 DFT 计算阐明 Co(0) 启动的 LLHT 反应机制,揭示 π-π 堆积、范德华作用及配体电子-位阻效应调控选择性的本质,为新型选择性硅氢化催化剂理性设计提供理论支撑。
这一成果近日发表于《中国化学》(Chinese Journal of Chemistry)杂志,文章第一作者为南开大学博士研究生刘祥雨。
文章信息
Cobalt-Catalyzed Hydrosilylation of 1,3-Enynes for the Synthesis of Conjugated Alkenylalkoxysilanes.
Xiang-Yu Liu, Jia-Lin Wang, Qiao Zhang, Ming-Yao Huang, and Shou-Fei Zhu*
Chin. J. Chem. 2026, 44, 2345-2353. DOI: 10.1002/cjoc.70606
